Fig. 1.1
Effects of Western diet consumption on the brain
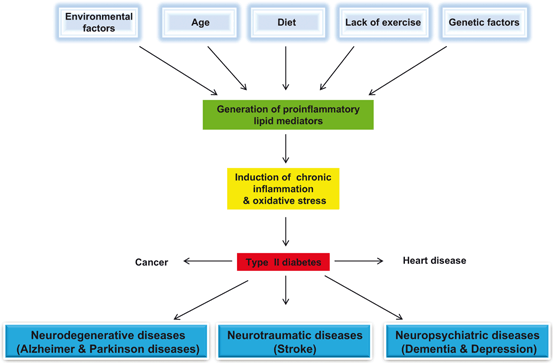
Fig. 1.2
Effect of age, diet, physical inactivity, and genetic and envioronmental factors of the pathogenesis of visceral and neurological disorders
As stated above, Western diet adversely impacts health of human brain. Long-term consumption of Western diet may not only induce mitochondrial abnormalities, facilitate the formation of protein aggregates (Aβ, neurofibrillary tangles, Lewy Bodies, and lipofuscin ceroid), reduce levels trophic factors, and promote the induction of neuroinflammation and oxidative stress (Fig. 1.1). These changes may cause functional impairments in the brain. The severity of brain impairments is highly dependent on the areas (brain region) and extent the damage to the brain. Western diet produces inflammation both in peripheral tissues and hypothalamus, an area critical for the maintenance of energy homeostasis (Thaler et al. 2012). Unlike inflammation in peripheral tissues, which develops as a consequence of obesity and insulin resistance after weeks to months, the onset of hypothalamic inflammation occurs both in rats and mice within 1 to 3 days after the start of high fat and high sugar diet and prior to substantial weight gain. Hypothalamic inflammation is accompanied by reactive gliosis involving both microglial and astroglial cell populations along with increase in markers of neuron injury (TNF-α, IL1-β, and IL-6) within a week. Although these responses temporarily subside due to the onset of neuroprotective mechanisms, which may initially limit the damage, but with continuation of high fat and high sugar diet uptake, inflammation and gliosis return permanently to the mediobasal hypothalamic region (Thaler and Schwartz 2010; Thaler et al. 2012). These observations on rodents are supported by MRI studies in humans, which indicate that there is an increase in gliosis in the mediobasal hypothalamus of obese humans. These findings collectively suggest that in both human and rodent consumption of high fat and high carbohydrate diet is associated with neuronal injury in the hypothalamus, an area of the brain involved in body weight control (Thaler et al. 2012). Several mechanisms have been proposed to explain high fat and high sugar-induced inflammation in both peripheral tissues and hypothalamus, including activation of TLR4, induction of endoplasmic reticulum stress, IKKβ/NF-κB signaling, and induction of SOCS3 along with other intracellular inflammatory signals associated with high levels of circulating saturated fatty acids (Thaler et al. 2012; Fessler et al. 2009; Zhang et al. 2008) that exacerbate the inflammatory response and facilitate insulin resistance. The relative contribution of these mechanisms in the onset and maintenance remains uncertain. However, much earlier onset of inflammation in hypothalamus relative to peripheral tissues suggests that different processes may be associated with the inflammation in peripheral tissues (Thaler and Schwartz 2010). In addition, consumption of Western diet is also associated with the alterations in hippocampal morphology/plasticity and impairment of cognitive function in normal rats (Granholm et al. 2008; Stranahan et al. 2008). Western diet-mediated changes in hippocampal morphology/plasticity are of considerable interest because this region is involved in learning and memory formation. Using normal C57BL/6 mice, it is also reported that a high-fat/high-cholesterol diet induces loss of working memory correlated with striking neuroinflammatory changes and increases in APP processing (Li et al. 2007; Thirumangalakudi et al. 2008).
1.2.1 High Fat Diet and Changes in BDNF Signaling
BDNF is a neurotrophic factor involved in the regulation of synaptic function and morphology (Horch 2004). High fat diet has ability to alter the level of BDNF. This growth factor is responsible for axon targeting, neuron growth, maturation of synapses during development, and synaptic plasticity (Kanoski et al. 2007). These changes in BDNF signaling are closely linked with deleterious changes in MAPK and CREB signaling, which are necessary for neuronal homeostasis (Kanoski et al. 2007). BDNF is expressed by the brain as well as in neuromuscular system where it protects neuromotor neurons from degradation (neuromotor neurons are critical element of our muscles. Without the neuromotor system, our muscles are like an engine without ignition). Collective evidence suggests that BDNF is actively involved in metabolic processes in both muscles and brain, and cross-connection between brain and muscles explains beneficial effects of exercise the brain.
1.2.2 High Fat Diet and Induction of Mitochondrial Abnormalities
Consumption of Western diet has been reported to down-regulate the expression of genes involved in oxidative phosphorylation and mitochondrial biogenesis (Nisoli et al. 2007). These processes are accompanied by an increase in fatty acid oxidation, as an alternative source for ATP generation (Nisoli et al. 2007). Long term upregulation of fatty oxidation in visceral tissues (muscle and liver) and in the brain may have adverse effects on cellular homeostasis through the elevation in oxidative stress and reduction in ATP production.
1.2.3 High Fat Diet and Induction of Oxidative Stress and Neuroinflammation
In the brain, Western diet produces oxidative stress, a condition that is caused by excessive formation and/or insufficient removal of highly reactive molecules called as reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS include superoxide anions (O2 •−), hydroxyl (•OH), alkoxyl (RO•-), and peroxyl radicals (ROO•), singlet oxygen, hydrogen peroxide (H2O2), and hydrochlorous acid (HOCl). RNS include free radicals such as NO and nitrogen dioxide (NO2 −), as well as non-radicals such as peroxynitrite (ONOO–), nitrous oxide (HNO2) and alkyl peroxynitrates (RONOO). The major sources of oxidative stress are mitochondrial respiratory chain, xanthine/xanthine oxidase, myeloperoxidase in cytoplasm, oxidation of ARA by cyclooxygenase (COX) and lipoxygenase (LOX) in cytoplasm, and NADPH oxidase in plasma membranes (Farooqui 2010) (Fig. 1.3). Under physiological conditions the antioxidant defense systems (superoxide dismutase, catalase, transferrin, and glutathione peroxide, glutathione, and vitamin C) within the body can easily neutralize the amount of ROS produced through three cellular antioxidative defense systems. However, disruption of tight control by high ROS levels results in the severe oxidative stress, which is closely associated with neural cell injury (Farooqui 2010). Low levels of ROS are not only needed for fundamental cellular functions, such as growth and adaptation responses, but also for optimal functioning of the immune system, which is involved in the defense against the penetrating microorganisms. However, generation of high ROS in neural cells facilitates the translocation of transcription factor (NF-κB) from cytoplasm to the nucleus where it facilitates the expression of proinflammatory enzymes (PLA2, COX, and nitric oxide synthase), proinflammatory cytokines and chemokines (TNF-α, IL-1β, COX-2, iNOS, VCAM-1 and ICAM-1 and adhesion molecules), growth factors, cell cycle regulatory molecules, and adhesion molecules (Fig. 1.3). Production of ROS is thought to exert a major influence on neural cell aging and tissue damage, particularly to cardiac and neural cells. ROS production tends to increase with age, as indicated by markers, such as lipid peroxidation and impaired antioxidant activity. Moreover, psychological stress and lifestyle factors such as smoking and sedentariness produce considerable impact on ROS production. ROS interact with proteins, nucleic acids, and lipids and produce damage to neural membranes. In an elderly population, ROS production is not only closely associated with poorer cognitive function and loss of ability to perform daily activities, but also with institutionalization, as well as depressive symptoms.
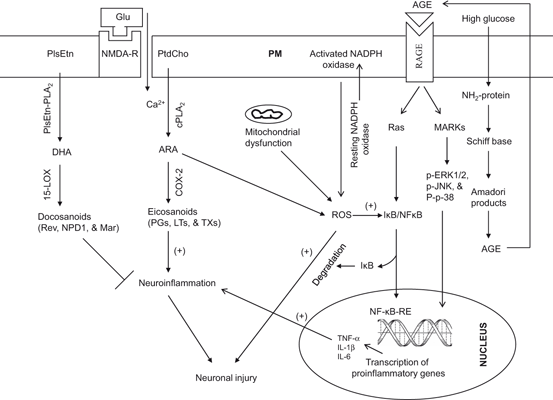
Fig. 1.3
Mechanisms contributing to oxidative stress and neuroinflammation in the brain. Plasma membrane (PM); N-methyl-D-aspartate receptor (NMDA-R); glutamate (Glu); phosphatidylcholine (PtdCho); ethanolamine plasmalogen (PlsEtn); plasmalogen-selective phospholipase A2 (PlsEtn-PLA 2); cytosolic phospholipase A2 (cPLA 2); cyclooxygenase (COX-2); 15-lipoxygenase (15-LOX); arachidonic acid (ARA); docosahexaenoic acid (DHA); reactive oxygen species (ROS); nuclear factor kappaB (NF-κB); nuclear factor kappaB response element (NF-κB-RE); inhibitory subunit of NF-κB (IκB); tumor necrosis factor-α (TNF-α); Interleukin-1 beta (IL-1β); Interleukin-6 (IL-6); prostaglandins (PGs); leukotrienes (LTs); thromboxanes (TXs); nicotinamide adenine dinucleotide phosphate oxidase (NADP oxidase); advanced glycation end-product (AGE); receptors for advanced glycation end-product (RAGE); mitogen-activated protein kinases (MAPK); and extracellular signal-regulated kinases (ERK)
In addition, elevation in glucose and plasma fatty acids due to long-term consumption of Western diet causes the activation of redox cycling of the copper-albumin complex and excessive lipid peroxidation, which lead to the accumulation of AGEs through the Maillard reaction (a non-enzymatic reaction between ketone group of the glucose molecule or aldehydes and the amino groups of proteins) (Brownlee 1995). Thus, AGEs are a group of highly stable crosslinked products that form through a series of reactions between glucose, proteins, lipids, and nucleotides. Humans are exposed to two main sources of AGEs: exogenous AGEs that are ingested in foods and endogenous AGEs that are formed in the body. The Western diet is rich in AGEs, which are formed when food is processed at elevated temperatures, such as during deep-frying, broiling, roasting, and grilling. In addition, high-temperature processing of certain processed foods such as pasteurized dairy products, cheeses, sausages, and processed meats; and commercial breakfast cereals result in generation of toxic products. While AGEs form during normal aging, their formation is accelerated in diabetes, hyperglycemia, and oxidative stress . All proteins are proned to AGE formation. Deleterious AGE accumulation occurs in tissues with low turnover, including the CNS. The binding of AGEs with AGE receptor (RAGE) not only results in the activation of the mitogen-activated protein kinases (MAPKs) and the phosphatidylinositol-3 kinase (PtdIns 3K) pathways, but also result in the stimulation of NADPH oxidases, generation of superoxide, and activation of NF-κB. This transcription factor migrates to the nucleus, where it increases the expression of inflammatory cytokines (TNF-α, IL-1β, IL-6, and vascular cell adhesion molecule 1) (Fig. 1.3) (Ahmed 2005; Farooqui 2013a). The other cell surface receptors for AGEs include dolichyl-diphosphooligosaccharide-protein glycosyltransferase (AGE-R1) (Li et al. 1996), protein kinase C substrate, 80KH phosphoprotein (AGE-R2) (Goh et al. 1996), galectin-3 (AGE-R3) (Vlassara et al. 1995), and macrophage scavenger receptors type I and II. RAGEs are also considered as pattern recognition receptors due to their ability to bind different AGEs. Thus, mounting evidence suggests that generation of AGEs through consumption of Western diet contributes to the induction and maintenance of oxidative stress and neuroinflammation (Pasinetti and Eberstein 2008). In addition, hypercholesterolemia can cause tau hyperphosphorylation in the brain, which has been shown to occur in apoE-deficient mice fed a high-cholesterol diet (Rahman et al. 2005). Collective evidence suggests that high fat and high carbohydrate diet produces neurodestructive effects in the brain (Zilberter 2011), where as low carbohydrate and high fat diet produces neuroprotective effects via induction of heat-shock proteins (Maalouf et al. 2009), growth factors (Maswood et al. 2004), and mitochondrial uncoupling proteins (Liu et al. 2006; Hipkiss 2008). It is becoming increasingly evident that high fat and high carbohydrate diet promotes AD type beta amyloidosis; conversely, intake of diet moderately enriched in fat and low in carbohydrate prevents AD type beta amyloidosis (Seneffs et al. 2011).
1.3 Effect of Mediterranean Diet on Human Brain
The Mediterranean diet is characterized by a high intake of vegetables, legumes, fruits, and cereals (largely unrefined); a moderate to high intake of fish; a low intake of saturated lipids but high intake of olive oil; a low to moderate intake of dairy products, mostly cheese and yogurt; a low intake of meat; and a modest intake of ethanol, mostly as red wine (Willett et al. 1995) . Mediterranean diet not only increases longevity by lowering cardiovascular disease, inhibiting cancer growth, but also protects the body from age-dependent cognitive decline (Fig. 1.4) (Perez-Jimenez et al. 2006; Lopez-Miranda et al. 2007). This is due to the presence of phytochemicals from fresh fruits and vegetables, sulfur compounds from garlic, resveratrols from red wine, and hydroxytyrosol and oleuropein from olives (Farooqui 2012). Olive oil contains oleic acid, a monounsaturated non essential fatty acid belonging to n-9 family of fatty acids. Olive oil not only provides the higher percent of energy but a lot of bioactive compounds (polyphenols) (Fig. 1.5) that promote beneficial effects on neurovascular and cardiovascular systems .
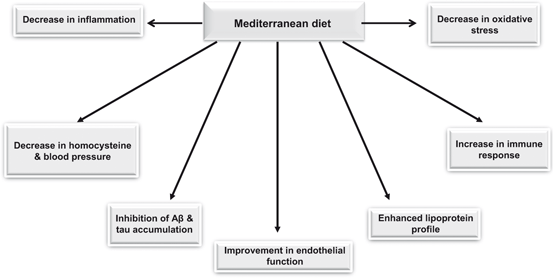
Fig. 1.4
Beneficial effects of mediterranean diet on human health
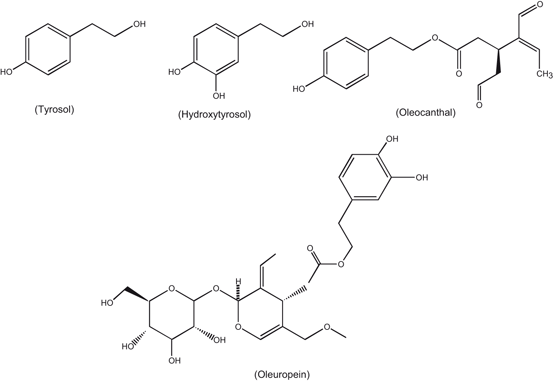
Fig. 1.5
Chemical structures of polyphenols found in olive oil
Polyphenol components of olive oil include tyrosol, hydroxytyrosol, oleocanthal, oleuropein (Fig. 1.5). These components produce protective effects against age-related cognitive decline and Alzheimer disease (Lopez-Miranda et al. 2007). Studies on the effect of olive oil, coconut oil, and butter on learning and memory in 11 month old SAMP8 mice for 6 weeks indicate that olive oil shows improvement in acquisition in the T-maze compared to coconut oil or butter treated mice. Olive oil increases brain glutathione levels suggesting reduced oxidative stress as a possible mechanism. In addition, effect of olive oil is accompanied by increase in glutathione reductase, superoxide dismutase activities, and decrease in tissue levels of 4-hydroxynoneal and 3-nitrotyrosine. These observations suggest that olive oil produces beneficial effects on learning and memory deficits found in aging and diseases, such as those related to the overproduction of amyloid-β protein (Farr et al. 2012). Oleic acid also inhibits gap junction permeability and increases glucose uptake in cultured rat astrocytes (Lavado et al. 1997). Although, the molecular mechanism of oleic acid-mediated inhibition of gap junction permeability is not fully understood, but it is proposed that oleic acid mediates its action through the stimulation of protein kinase C (PKC) activity (Khan et al. 1992), a process that increases the rate of cell proliferation by enhancing glucose uptake . In addition, during brain development, neurons use oleic acid for the synthesis of glycerophospholipids and incorporate it into growth cones (Tabernero et al. 2001). Oleic acid promotes axonal growth, neuronal clustering, and expression of the axonal growth-associated protein-43 (GAP-43). The effect of oleic acid on GAP-43 synthesis is also mediated through the activation of PKC, since it can be blocked by inhibitors of this kinase, such as H-7, polymyxin or sphingosine (Granda et al. 2000). The expression of GAP-43 is significantly increased in neurons co-cultured with astrocytes in the presence of albumin. The effect of albumin can be blocked either by retarding traffic to the endoplasmic reticulum or adding albumin-oleic acid complex. In addition, oleic acid also mediates the expression of microtubule associated protein-2 (MAP-2), a marker of dendritic differentiation (Rodríguez-Rodríguez et al. 2004) . The time course of MAP-2 expression during brain development coincides with that of stearoyl-CoA desaturase, the limiting enzyme of oleic acid synthesis, indicating that both phenomena coincide during development. It is proposed that the effect of oleic acid on MAP-2 expression is most probably independent of autocrine factors synthesized by neurons (Rodríguez-Rodríguez et al. 2004) and exogenous or endogenous oleic acid by astrocytes exerts its neurotrophic effect through a protein kinase C-dependent mechanism. This effect can be prevented by sphingosine or two myristoylated peptide inhibitors of protein kinase C (Rodríguez-Rodríguez et al. 2004). Collectively, these studies indicate that during brain development, the presence of albumin plays an important role by triggering the synthesis and release of oleic acid by astrocytes, which induces neuronal differentiation through its interactions with transcription factor NeuroD2 (Tabernero et al. 2001; Taberenero et al. 2002; Rodríguez-Rodríguez et al. 2004) .
Polyphenol components of olive oil have been reported to produce beneficial effects in animal models of stroke and Alzheimer disease (Bu et al. 2007; Rigacci et al. 2010; Rigacci et al. 2011). In an animal model of stroke, tyrosol (30 mg/kg) produces a dose-dependent neuroprotective effects (Bu et al. 2007). In addition, rotarod beam balance, and foot fault tests, tyrosol exhibits protective effects against the sensory motor dysfunction supporting the view that tyrosol is a neuroprotective agent (Perez-Jimenez et al. 2006, 2007; Bu et al. 2007). Similarly, oleuropein, a main constituent of the olive tree leaves and unprocessed olive, interferes with the in vitro aggregation of human amylin and Aβ42 resulting in inhibiting the appearance of toxic oligomers and promoting peptide aggregation into aggregates devoid of cytotoxicity (Rigacci et al. 2010; Rigacci et al. 2011) . It is also shown that oleuropein aglycon is maximally effective when present at the beginning of the aggregation process. Furthermore, when added to preformed fibrils, it does not induce the release of toxic oligomers but, rather, neutralizes any residual toxicity possibly arising from the residual presence of traces of soluble oligomers and other toxic aggregates (Rigacci et al. 2011). In addition, it is also reported that oleuropein aglycone also inhibits the aggregation of Tau protein, another protein closely associated with the pathogenesis of AD (Daccache et al. 2011). It should also be noted that Mediterranean diet, which is known for its high content of nitrate-rich leafy green vegetables, lowers the blood pressure of healthy volunteers (Lundberg et al. 2006; Larsen et al. 2006). The nitrate/nitrite/NO pathway through oral ingestion relies on a symbiotic relationship with natural oral flora. Nitrate is concentrated within the salivary glands and salivary bacteria. It reduces nitrate to nitrite in the oral cavity (Duncan et al. 1995). Upon arrival in the stomach nitrite is reduced to NO by protonation, which is facilitated by low pH of the stomach (Benjamin et al. 1994) . NO then may act locally by enhancing mucosal blood flow to the stomach (Benjamin et al. 1994; Björn et al. 2004). Absorbed nitrite can enter blood stream from the stomach (Miyoshi et al. 2003). Due to its relative stability, nitrite then has the ability to circulate to other areas in the body and undergoes reduction to NO under acidic and hypoxic conditions (Cosby et al. 2003). Acting in this manner, circulating nitrite acts as a “storage pool” for NO within the body (Dejam et al. 2004; Madigan and Zuckerbraun 2013). Collective evidence suggests that the Mediterranean diet provides protection against cognitive decline in older individuals since it combines several foods and nutrients (n-3 fatty acid in fish, monounsaturated fatty acids, vitamins B12 and folate, antioxidants in olive oil, nitrate in green vegetables, and resveratrol), which are protective against cognitive dysfunction, dementia, and neurodegeneration (Scarmeas et al. 2006) .
1.4 Effect of Ketogenic Diet on Human Brain
The ketogenic diet is a high-fat, low-carbohydrate and low protein diet that alters metabolism by increasing the level of ketone bodies in the blood . Fatty acids are an obligatory source of cellular energy production not only by peripheral tissues and also by the brain. Fatty acids are transported across the blood brain barrier through diffusion and transport mechanisms. Diffusion involves a free fatty acid flip-flop process, where as transport mechanism involves fatty acid transporters [fatty acid transport proteins (FATPs), fatty acid translocase (FAT/CD36), and fatty-acid-binding proteins (FABPs)]. Consumption of the ketogenic diet is characterized by increase in circulating levels of the ketone bodies (acetoacetate, β-hydroxybutyrate and acetone), which are largely produced by the liver mitochondria (Fig. 1.6). During high rates of fatty acid oxidation, large amounts of acetyl-CoA are generated, which exceed the capacity of the TCA cycle and lead to the synthesis of ketone bodies within liver mitochondria. Plasma levels of ketone bodies rise, with acetoacetate and β-hydroxybutyrate increasing 3–4-fold from basal levels of 100 and 200 µmol/l, respectively (Musa-Veloso et al. 2002). The accumulation of ketone bodies in the bloodstream causes ketosis, a survival mechanism activated during prolonged fasting, starvation or lack of carbohydrate ingestion. During prolonged ketosis, the brain is capable of utilizing ketone bodies as an alternate fuel, thus reducing its requirement for glucose (LaManna et al. 2009). The ketone bodies are oxidized, releasing acetyl-CoA, which enters the TCA cycle for energy production. Although, ketogenic diet has been used for the treatment of epilepsy for more than 90 years (Wilder 1921), but recent studies have indicated that ketogenic diet can provide symptomatic and disease-modifying activity in a broad range of neurodegenerative disorders including animal models of Alzheimer disease and Parkinson disease, stroke, multiple sclerosis, amyotrophic lateral sclerosis, and traumatic brain injury (Gasior et al. 2006; Van der Auwera et al. 2005; Zhao et al. 2006; Maalouf et al. 2009). Metabolic changes likely related to anticonvulsant properties of ketogenic diet include—but are not limited to—ketosis, reduction in glucose levels, elevation in fatty acid levels, and enhancement in bioenergetic reserves. Ketogenic diet not only modulates ATP-sensitive potassium (KATP) channels and enhances purinergic (i.e., adenosine) and GABAergic neurotransmission, but also increases BDNF expression and attenuates neuroinflammation (Politi et al. 2011). In addition, it also promotes an expansion in bioenergetic reserves and facilitates the stabilization of the neuronal membrane potential through improved mitochondrial function (Fig. 1.7). These processes exert neuroprotective and antiepileptogenic properties, heightening the clinical potential of the ketogenic diet as a disease-modifying intervention (Politi et al. 2011). The consumption of ketogenic diet not only produces reduction in blood glucose, decreases in ROS , and increases in glutathione peroxidase activity in the hippocampus but also increases uncoupling proteins expression and alterations in the expression of genes associated with oxidative stress (Sullivan et al. 2004; Ziegler et al. 2003; Bough et al. 2006). Furthermore, ketogenic diet also stimulates mitochondrial biogenesis and increases cerebral ATP and phosphocreatine concentrations, suggesting increase in metabolic efficiency (Fig. 1.7) (Bough et al. 2006). In experimental autoimmune encephalomyelitis model (EAE), ketogenic diet improves motor disability as well as CA1 hippocampal synaptic plasticity (long-term potentiation) and spatial learning and memory (assessed with the Morris Water Maze) (Kim do et al. 2012). Moreover, hippocampal atrophy and periventricular lesions in EAE mice can be reversed in ketogenic diet-treated EAE mice. In EAE mice ketogenic diet suppresses the increase in expression of inflammatory cytokines and chemokines, as well as production of ROS . Based on these studies, it is suggested that neuroinflammation in EAE mice is associated with impaired spatial learning and memory function, and that ketogenic diet treatment can induce neuroprotection through the attenuation of the robust immune response and suppression of oxidative and neuroinflammation in these animals (Kim do et al. 2012). Ketogenic diet produces neuroprotective effects by providing an alternative source of energy in states of metabolic stress. In fact, β-hydroxybutyrate may provide a more efficient source of energy for brain per unit oxygen than glucose (Veech et al. 2001). Recent microarray studies have indicated that the ketogenic diet produces a coordinated upregulation of hippocampal genes encoding energy metabolism and mitochondrial enzymes (Bough et al. 2006). Ketogenic diet also increases production of specific mitochondrial uncoupling proteins (UCPs) (Sullivan et al. 2004). For example, in mice fed with ketogenic diet, UCP2, UCP4, and UCP5 are increased, particularly in the dentate gyrus. UCPs serve to dissipate the mitochondrial membrane potential, which, in turn, decreases the formation of ROS (Freeman et al. 2006). Accumulating evidence suggests that consumption of ketogenic diet treatment not only produces an increase in ketone body production by the liver, but also reduces blood glucose levels. The elevation of ketones is largely a consequence of fatty acid oxidation. Polyunsaturated fatty acids (PUFAs) themselves regulate neuronal membrane excitability by blocking voltage-gated sodium and calcium channels (Voskuyl and Vreugdenhil 2001), reducing inflammation through activation of peroxisome proliferator-activated receptors (PPARs) (Jeong et al. 2011), or inducing expression of mitochondrial uncoupling proteins which reduce ROS production (Kim do and Rho 2008). Ketone bodies produce neuroprotective effects by raising ATP levels and reducing ROS production through enhanced NADH oxidation and inhibition of mitochondrial permeability transition (mPT) (Kim do et al. 2011). Ketogenic diet initially produces mild oxidative and electrophilic stress which may systemically activate the Nrf2 pathway via redox signaling leading to chronic cellular adaptation, induction of protective proteins, and improvement of the mitochondrial redox state (Milder et al. 2010). In addition, ketogenic diet not only improves bioenergetics by stimulating the mitochondrial biogenesis, but also by stabilizing synaptic function (Bough et al. 2006). Another important feature of the ketogenic diet is the decrease in glycolytic flux resulting in decrease in blood glucose. It is interesting to note that 2-Deoxy-d-glucose (2DG), an analog of glucose that inhibits phosphoglucose isomerase and hence inhibits glycolysis, blocks epileptogenesis in the rat kindling model by decreasing the expression of BDNF and its principal receptor, tyrosine kinase B (TrkB; Garriga-Canut et al. 2006) supporting the view that ketogenic diet produces beneficial effects in neurological disorders .
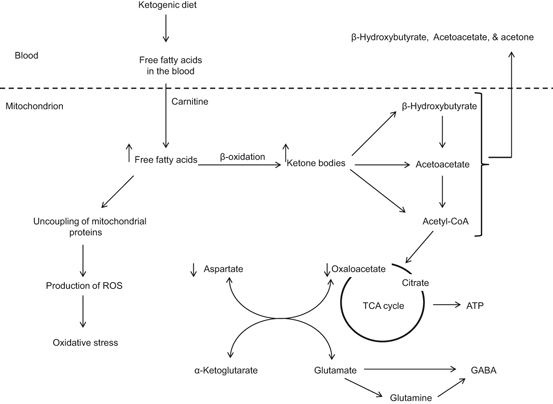
Fig. 1.6
Production of ketone bodies by ketogenic diet. γ-aminobutyric acid (GABA); reactive oxygen species (ROS); and tricarboxylic acid cycle (TCA)
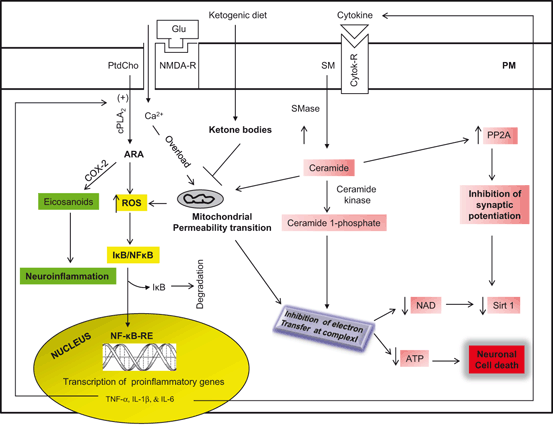
Fig. 1.7
Molecular mechanism associated with neuroprotective effects of ketogenic diet. Plasma membrane (PM); N-methyl-D-aspartate receptor (NMDA-R); glutamate (Glu); phosphatidylcholine (PtdCho);cytosolic phospholipase A2 (cPLA2); cyclooxygenase (COX-2); 15-lipoxygenase (15-LOX); arachidonic acid (ARA); reactive oxygen species (ROS); nuclear factor kappaB (NF-κB); nuclear factor kappaB response element (NF-κB-RE); inhibitory subunit of NFκB (IκB); tumor necrosis factor-α (TNF-α); Interleukin-1 beta (IL-1β); Interleukin-6 (IL-6); sphingomyelin (SM); sphingomyelinase (SMase); sirtuin 1 (Sirt 1); cytokine receptor (cytokine-R); and apoptotic enzyme phosphatase 2A (PP2A)
1.5 Calorie Restriction and its Effects on the Brain
Calorie restriction refers to a reduced calorie intake (a reduction of food intake by 20 to 40 %) without malnutrition. Beneficial effects of calorie restriction on the brain are mediated through the activation of adaptive cellular stress responses, in a process called hormesis (Cornelius et al. 2013; Mattson 2008a, b). Hormesis is defined as an adaptive response of cells and organisms to a moderate (usually intermittent) stress. At the molecular level, calorie restriction reduces the generation of ROS and nitrogen species in mitochondria during the food metabolism. The molecular mechanism involved in this process is not fully understood. However, it is proposed that under calorie restriction, sirtuin (SIRT3) deacetylates and subsequently activates SOD2, leading to reduction in oxidative stress (Chen et al. 2011). This suggestion is supported by studies on the protective effects of calorie restriction on oxidative stress in mice lacking SIRT3 (Qiu et el. 2010). Moreover, beyond the activation of SOD2, SIRT3 also mediates other antioxidant functions in mitochondria. For instance, a comparative study of Sirt3 wild-type and knockout mice has shown that SIRT3 induces the beneficial effects of calorie restriction on age-related hearing loss and prevents the accumulation of ROS under calorie restriction in cochlea cells through the deacetylation and activation of isocitrate dehydrogenase 2 (IDH2) (Someya et al. 2010). SIRT3 also activates glutamate dehydrogenase (GDH) (Giralt and Villarroya 2012). Activation of IDH2 and GDH indirectly stimulates the production of reduced glutathione, the cofactor used by glutathione peroxidase (GPx) to detoxify ROS (Giralt and Villarroya 2012; Bell and Guarente 2011).
Calorie restriction mediates a mild stress on cells through the activation of stress response pathways (Fig. 1.8) involving transcription factors, CREB, Nrf-2, AP-1, and NF-κB (Mattson and Cheng 2006; Kim et al. 2002). The cellular stress can be exerted by a combination of the direct consequences of reduced energy intake and an increase in the activity of neuronal circuits. In the brain, calorie restriction is accompanied by the upregulation of adaptive stress response proteins in neurons (Martin et al. 2006). Reducing energy intake by controlled caloric restriction or intermittent fasting not only increases lifespan (by 20–40 %) and protects neural cells against neurological disorders , but also by enhances cellular stress resistances. Calorie restriction also decreases age-related learning and memory impairments in animals and human (Witte et al. 2009), probably through higher expression of an NMDA-receptor subunit in the hippocampus. In addition, calorie restriction not only attenuates age-related brain atrophy in monkeys (Colman et al. 2009), but also stabilizes the expression of synaptic protein expression to avoid aging-related changes (Mladenovic et al. 2010). Many mutations that extend life span decrease activity of nutrient-signaling pathways, such as the insulin-like growth factor (IGF)/insulin, mammalian target of rapamycin (mTOR) pathways, AMPK, and PGC-1α signaling pathways supporting the view that these pathways may induce a physiological state similar to that resulting from periods of food shortage (Wang et al. 2010a; Bonda et al. 2011). Furthermore, it is also reported that Sirt1 (a mammalian member of the sirtuin gene family) contributes to the beneficial impact of caloric restriction , which may be mediated, in part, through mechanisms involving the regulation of cellular metabolism, and inflammatory and antioxidant responses (Furuya et al. 2012). Consumption of high fat and high carbohydrate diet produces AD type Beta-amyloidosis, where as dietary restriction based on reduced carbohydrate intake prevents AD type Beta-amyloidosis (Seneff et al. 2011).
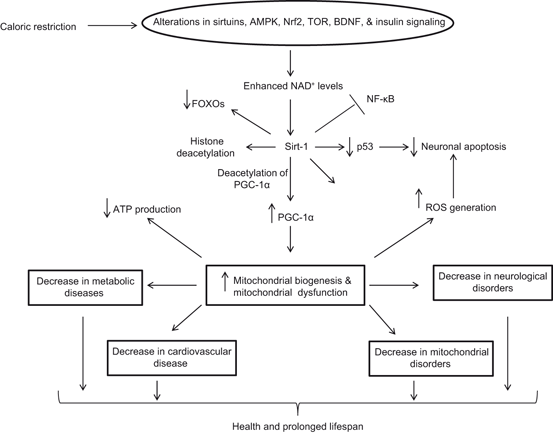
Fig. 1.8
Effect of calorie restriction on health and longevity. Nuclear factor kappaB (NF-κB); Forkhead box protein O (FOXO); NF-E2-Related Factor 2 (Nrf2); AMP-activated protein kinase (AMPK); brain-derived neurotrophic factor (BDNF); sirtuin-1 (Sirt-1); PPARγ coactivator-1α (PGC-1α); and reactive oxygen species (ROS)
Some specific dietary components have been reported to exert health benefits by inducing adaptive cellular stress responses. Recent studies have indicated that several phytochemicals exhibit biphasic dose responses in neural cells with low concentrations activating signaling pathways that result in increased expression of genes encoding cytoprotective proteins such as antioxidant enzymes, protein chaperones, growth factors and mitochondrial proteins (Mattson 2008a, 2008b). Examples include: activation of the Nrf-2—ARE pathway by sulforaphane and curcumin; activation of TRP ion channels by allicin and capsaicin; and activation of sirtuin-1 by resveratrol (Mattson 2008a, 2008b). Thus, phytochemicals containing foods such as flavonoids in blueberries, catechins in green tea, and curcumin in turmeric increase multiple parameters of hippocampal synaptic plasticity, and these parameters correlated with improvements in spatial memory (Casadesus et al. 2004; Farooqui 2012).
Mitochondria are a major source of ROS in the neural cells and dietary restriction directly reduce the generation of ROS by minimizing proton leakage (Hagopian et al. 2005; Civitarese et al. 2007; Bevilacqua et al. 2004; Bevilacqua et al. 2005). Conversely, a high fat diet is known to stimulate the expression of NF-kappa B resulting in neuroinflammation (Liao et al. 1993), which may utimately lead to increase in cellular stress. ROS-mediated damage is ameliorated by a antioxidant systems acting at the cellular level to protect the integrity of biomolecules from oxidative damage (Bayir and Kagan 2008), Emerging evidences, both in in vitro and in vitro studies indicate that increase in ATP expenditure through calorie restriction exert beneficial effects on the brain primarily through improvement in antioxidant systems, upregulation of growth factors (BDNF, NGF, and IGF) and cytoprotective proteins, such as heat shock protein 72 (Hsp 72) (Hofer et al. 2008), and mild uncoupling in the passage of protons through the inner mitochondrial membrane from mitochondrial phosphorylation (Liu et al. 2006). This uncoupling is partly mediated by the uncoupling proteins (UCP), whose levels are increased by calorie restriction in various tissues, including the neuron-specific UCP4 (Liu et al. 2006). As a consequence, these mechanisms are involved in enhancing the efficiency of ROS neutralization so as to reduce the risk factor of age-related oxidative damage and chronic neurological diseases (Fig. 1.9). Collectively, these studies indicate that calorie restriction not only decreases in ROS generation and neuroinflammation, but also increases BDNF production, and enhances immune responses (Fig. 1.10). Paradoxically, caloric restriction induces mitochondrial proliferation in rodents, and has no affect mitochondrial oxygen consumption (Lambert et al. 2004; Nisoli et al. 2005) supporting the view that calorie restriction improves whole body energy efficiency by inducing the biogenesis of mitochondria that utilize less oxygen and produce less ROS.
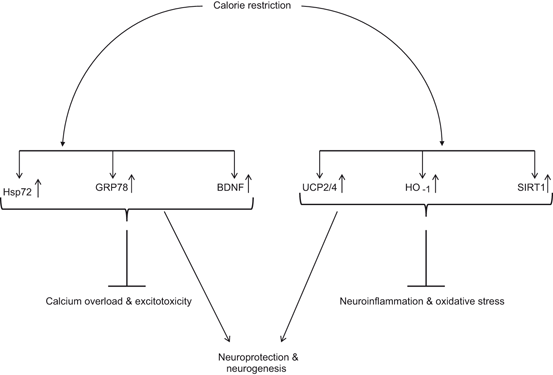
Fig. 1.9
Effect of calorie restriction on factors and proteins that increase neuronal survival and neurogenesis. Heat shock protein 70 (Hsp70); 78 kDa glucose-regulated protein (GRP78); uncoupling proteins (UCP); Brain-derived neurotrophic factor (BDNF); hemeoxygenase-1 (HO-1); and sirtuin (SIRT1)
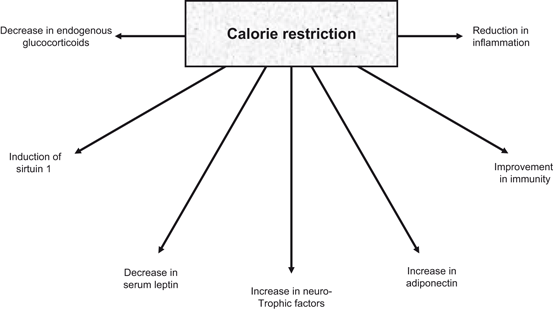
Fig. 1.10
Neurochemical consequences of calorie restriction in the brain
Calorie restriction affects specific regions of aging brain and this process very much dependent on the neuronal and synaptic substrates of that specific area and its neuronal circuits (Mora et al. 2007). For example, it is well known that the gray matter volume in the caudate nucleus decreases with age in control animals, but is preserved in calorie-restricted monkeys (Colman et al. 2009). In contrast, other areas of the monkey brain, including the frontal and temporal cortex are characterized by a significant reduction in gray matter volume that is not decreased by a reduction in food intake (Colman et al. 2009). In addition, calorie restriction enhances BDNF levels in several areas of the brain, particularly the hippocampus (Fig. 1.10) (Lee et al. 2002). Calorie restriction not only mediates protective effects in animal models of Alzheimer and Parkinson diseases, but also induces neuroprotective changes in animal models of other neurodegenerative disorders (Masswood et al. 2004; Love 2005). For example, in mouse models of Alzheimer disease, calorie restriction reverses deficits in learning and memory typically found in these animals (Halagappa et al. 2007). Similarly, motor impairment associated with a monkey model of Parkinson disease can be attenuated by calorie restriction (Love 2005). Although, molecular mechanisms of these processes are not clear at the present time, but contribution of neurotrophic factors in calorie restriction-mediated neuroprotective effects has been proposed. as well as other proteins and enzymes on these protective effects of caloric restriction has been suggested (Stranahan and Mattson 2012).
Calorie restriction lowers plasma insulin levels and mediates greater sensitivity to insulin by decreasing leptin and increasing adiponectin, lowing body temperatures; reducing cholesterol, triglycerides and blood pressure, and increasing levels of human growth hormone, which is commonly known as “the fitness hormone”. This hormone plays an important role in maintaining health, fitness and longevity, including promotion of muscle growth, and boosting fat loss by revving up your metabolism. The fact that it helps build muscle while simultaneously promoting fat loss explains why human growth hormone promotes weight loss without sacrificing muscle mass and why even athletes can benefit from the practice (as long as they don’t over-train and are careful about their nutrition). Calorie restriction also elevates HDL and slows age related decline in circulating levels of dehydroepiandrosterone sulfate. As stated above, calorie restriction mediates the synthesis of cellular stress-response stimulating proteins (neurotrophic factors, neurotransmitter receptors, Hsps, and mitochondrial biosynthesis regulators) and enhances neuronal plasticity and resist oxidative and metabolic insults (Lee et al. 2000; Fontan-Lozano et al. 2008). Calorie restriction also regulates intracellular redox status through its antioxidative actions. Based on above discussion, it is proposed that calorie restriction as well as an active lifestyle from childhood to old age may facilitate prevention or postponement of the onset of neurological disorders .
1.6 Effect of Exercise on Signaling in Visceral Organs
Ancient men were hunters-gatherers, who used to travel long distances to collect food, avoid threats wild animals, and seek shelters. In contrast, modern western men have transformed their lifestyle to minimize environmental threats and stresses that were faced by our ancestors. Unlike our ancestors, modern western men have almost unlimited supplies of foods containing high level of saturated fats, vegetable oils (n-6 fatty acids) , salt, and refined sugars along with lack of consumption of food from plants (fruits, nuts and vegetables) and sea foods, which are enriched in n-3 fatty acids . Long term consumption of Western diet potentiates the appearance of obesogenic changes, which initiate the pathogenesis of metabolic diseases (diabetes, metabolic syndrome, and vascular diseases). Metabolic diseases are important risk factors for neurotraumatic (stroke) , neurodegenerative (Alzheimer disease) , and neuropsychiatric (depression) diseases (Nunn et al. 2010; Farooqui 2013a).
To optimize energy production and minimize oxidative damage at the subcellular and molecular levels, mitochondria are involved in dynamic network exchange through fission and fusion, which identifies damaged mitochondrial and tags them for mitophagy (Schafer and Reichert 2009). Impairments in mechanisms of mitochondrial quality control during aging is associated with the age-related increase in tissue oxidative damage and functional decline (Weber and Reichert 2010; Gottlieb and Carreira 2010). Exercise is an effective nonphramacological and physiological intervention that counteracts age-related mitochondrial dysfunction through simultaneous improvement of mitochondrial biogenesis and quality control, including up-regulation of mitophagy (Bori et al. 2012; Lanza and Sreekumaran 2012; Guo et al. 2012). Accumulating evidence suggests that exercise not only triggers mitochondrial biogenesis, a decline of which is common in aging, but also reverses significant age-associated declines in mitochondrial density, and stops aging in its tracks (Kottai et al. 2012). The main regulator of mitochondrial biogenesis is the peroxisome proliferator-activated receptor gamma coactivator (PGC) family of transcriptional activators. PGC family consists of PGC-1α, PGC-1β, and PGC-related coactivator (PRC) (Lin et al. 2005). PGC-1α plays a role in the activation of nuclear respiratory factor 2 and together they co-activate nuclear respiratory factor 1. Consequently, nuclear respiratory factor 1 activates Tfam, which is important for mitochondrial DNA (mtDNA) transcription, translation, and repair. Thus, PGC-1 family coactivators act as mediators between the environment and the transcriptional machinery regulating the biogenesis of mitochondria (Scarpulla 2008). Exercise also induces cardiorespiratory and muscular fitness by increasing energy consumption, improving insulin sensitivity, increasing blood flow, strengthening the immune system, reducing inflammation , promoting sleep, and controlling weight (Fig. 1.11). The molecular mechanisms underlying above mentioned processes are not fully understood. However, it is becoming increasingly evident that exercise not only improves the dyslipidemic profile by raising high density lipoprotein-cholesterol and lowering triglycerides in the body (Lakka and Laaksonen 2007), but also increases expression of GLUT4 and other proteins involved in insulin signaling and glucose metabolism (Houmard et al. 1993). In addition, exercise modulates the expression of certain genes and proteins, such as Hsps in the skeletal muscle. These proteins not only facilitate repair from injury, and provide protection against future insults, but also promote permanent beneficial adaptations, which may include increase in activators of mitochondrial biogenesis, such as PGC-1α and increase in activity of oxidative enzymes such as citrate synthase and succinate dehydrogenase (Carter et al. 2001; Fernström et al. 2004). In addition, Hsp peptides promote the production of anti-inflammatory cytokines, indicating immunoregulatory potential of these proteins. Therefore, the presence of immune responses to Hsps in inflammatory diseases is considered as an attempt of the immune system to correct the inflammatory condition (Lakka and Laaksonen 2007; Morton et al. 2009). It is also proposed that exercise boosts the production of human growth hormone and blocks maladapted pathways, which may not only provide protection against protein misfolding diseases, but also promote preservation of muscle function during aging (Morton et al. 2009). Recent studies have indicated that human genome contains genes controlling either the motivation or ability to exercise (Good et al. 2008). Polymorphism in these genes may lead to individuals with overweight and reduced physical activity (obese phenotype). One of these genes is called as nescient helix-loop-helix 2 (NHLH2, human; Nhlh2, mouse). NHLH2 is a basic helix-loop-helix transcription factor that controls body weight through control of physical activity levels. When Nhlh2 is deleted in mice (N2KO mice), adult-onset obesity occurs (Good et al. 1997; Coyle et al. 2002). Overeating does not produce obesity in N2KO mice, but rather reduces spontaneous physical activity levels (Coyle et al. 2002) suggesting that functional inactivation of the Nhlh2 gene may lead to decrease in the performance of daily physical activity and that this may be one of the contributing factors in the onset of obesity in N2KO mice (Good et al. 2008). Genetic studies have indicated that in humans that body weight differences can be linked to 11 single-gene mutations and to more than 600 other genes that act together to influence various aspects of body weight control (Rankinen et al. 2006; Fox et al. 2007). Possible phenotypic influences are likely to include both the motivation and ability to perform moderate physical activity on a weekly basis (Good et al. 2008).
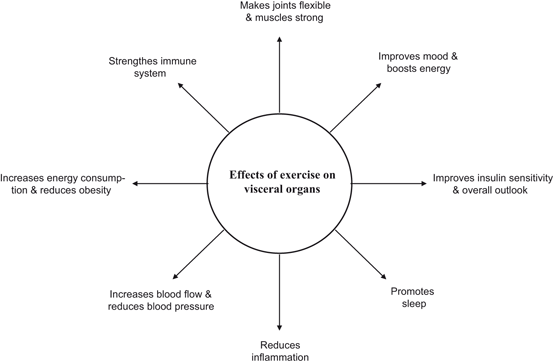
Fig. 1.11
Beneficial effects of exercise on visceral organs
Exercise blunts Toll-like receptor-4 (TLR-4) signaling and induces improvement in post-receptor insulin action (Oliviera et al. 2011). It not only reduces protein tyrosine kinase phosphatase 1B activity and insulin receptor substrate 1 serine phosphorylation, but also reduces c-jun N-terminal kinase activity in the muscle of diet-induced obese rats (Da Silva et al. 2010). These processes not only result in strengthening of bones and muscles, but also in increasing the longevity. In contrast, a sedentary lifestyle leads in a positive energy balance, which results in accumulation of body fat. Under these circumstances, adipose tissue initiates the expression and secretion of adipocytokines (tumor necrosis factor alpha (TNF-α), plasminogen activator inhibitor, and resistin) into the circulation. Adipocytokines not only induce low grade inflammation , but also impair glucose transport via the inhibition of insulin signal transduction. Insulin-induced activation of the insulin receptor (IR), phosphatidylinositol 3-kinase (PtdIns 3K), and Akt is retarded by the activation of IκB kinase (IKK) and degradation of IκB in the muscle tissue (De Alvaro et al. 2004). Furthermore, IKK-β silencing prevents TNF-α-induced impairments in insulin action on Akt phosphorylation and glucose uptake (Austin et al. 2008). Sedentary lifestyle results in atrophy of skeletal muscles (Zurlo et al. 1990) resulting in the loss of muscle fibers (Larsson and Ansved 1995) not only due to apoptotic cell death in the muscle (Sakuma and Yamaguchi 2010, 2012), but due to decrease in differentiation of satellite cells (Conboy and Rando 2005) as well as decrease in protein synthesis and increase in protein degradation (Combaret et al. 2009). The activity of enzymes associated with aerobic metabolism and glucose uptake in muscle is also decreased in the sedentary life style specifically in the middle age. In contrast, daily exercise produces a persistent increase in insulin action in the skeletal muscle of insulin-resistant individuals. The molecular mechanism(s) associated with increased glucose uptake with exercise have been attributed to the increased expression and/or activity of key signaling proteins involved in the regulation of glucose uptake and metabolism in skeletal muscle, up-regulation of genes encoding energy metabolism and increase in oxidative phosphorylation (Short 2009). At the molecular level exercise-mediated improvements in insulin sensitivity are related to changes in the expression of AMP-activated protein kinase (AMPK) and protein kinase B (Akt) substrate AS160 (Hawley and Lessard 2008). In addition, exercise increases oxidative capacity of the skeletal muscle by up-regulating lipid oxidation and the expression of proteins involved in mitochondrial biogenesis. This may be another mechanism by which exercise improves insulin sensitivity (Hawley and Lessard 2008). Accumulating evidence suggests that exercise, inflammation and immunity are tightly linked in a complex way (Febbraio 2007). Regular, moderate exercise reduces systemic inflammation (Gleeson 2007). Very little is known about mediators of this beneficial effect of exercise. However, it is reported that exercise increases the release of epinephrine, cortisol, growth hormone, prolactin and other factors that have immunomodulatory effects (Nieman 2003). Furthermore, exercise decreases the expression of Toll-like receptor on monocytes (Gleeson et al. 2006). In contrast to the reduction of chronic inflammation by moderate and regular exercise, prolonged high intensity exercise not only increases cortisol and systemic inflammation, but also in elevates the risk of infection (Gleeson 2007). In fact, high intensity exercise produces a transient exercise-induced immunodepression in athletes (Gleeson et al. 2004).
Exercise also produces beneficial effects on the vasculature. It increases vasodilation and improves vascular compliance, which are likely a result of shear stress and cell stretch on both the endothelium and underlying smooth muscle (Green et al. 2011; Wang et al. 2007). Exercise induces vasodilation through nitric oxide production and protects the vasculature through a number of mechanisms including reduction in inflammation (Warburton et al. 2006; Gleeson et al. 2011). Short-term exercise not only decreases the levels of TNF-α, IL-6, and plasminogen activator inhibitor-1 (PAI-1) (Izadpanah et al. 2012), but also reduces the expression of cell adhesion molecules (Saetre et al. 2011). These processes protect against media-intimal hyperplasia (Pahkala et al. 2011) and smooth muscle cell hypertrophy (Pahkala et al. 2011), and strengthens the endothelial barrier (Ding et al. 2006). However, intense exercise increases cortisol (Van Bruggen et al. 2011; Walsh et al. 2011), C-reactive protein (Mendham et al. 2011), and other proinflammatory cytokines , which may produce harmful effects, such as weight gain (Scott et al. 2011). Studies on human blood have indicated that moderate exercise increases the expression of inducible polyubiquitin genes. The molecular cascades of gene expression induced by exercise are multiple and document exercise as a hormetic agent with therapeutic potential for benefit and energy upregulation. Cross talk between major caloric restriction hormesis induced pathways especially AMPK/PPARδ and IGF1 reveal the linking of caloric restriction and exercise mimetics, contributing to the beneficial effects of exercise (Narkar et al. 2008) . Collective evidence suggests that moderate exercise produces human health benefits by improving cardiovascular and cerebrovascular systems, reducing osteoporosis/fracture risk and age-related sarcopenia, improving psychological effect, and inducing anti-inflammatory effect (Petersen and Petersen 2005). However, signal transduction processes and mediators of above mentioned effects are unresolved. A number of mechanisms have been identified. These include the release of adrenaline, cortisol, growth hormone, prolactin and other factors that have immunomodulatory effects (Handschin and Spiegelman 2008).
1.7 Effect of Exercise on Signaling in the Brain
Exercise is a general term, which is used to describe a physical activity that is planned, structured, and repetitive for the purpose of conditioning the body. In contrast, physical activity refers to “bodily movement produced by the contraction of skeletal muscle that requires energy expenditure in excess of resting energy expenditure.” Physical activity includes a broad range of occupational, leisure, and daily activities (Zanuso et al. 2010). Like skeletal muscles, in brain mitochondria undergo fission and fusion under physiologic conditions not only to maintain optimal morphological characteristics necessary to match ATP production to cellular needs, but also for regulation of calcium homeostasis and control Fe/S cluster biogenesis. Maintaining a balance between fission and fusion is important in neurons because of high neuronal energy demand and long mitochondrial transport distances especially in motor neurons (Chen and Chan 2010; Uo et al. 2009). Therefore, in neural cells the balance shifts toward fission compared with non-neural cells in order to maintain small, highly motile mitochondria consistent with need (Uo et al. 2009). The ability to learn new tasks decreases with age due to reduction in synaptic contacts, synaptic strength, and neural plasticity. In addition, hippocampal neurogenesis is also diminished with aging (Erickson et al. 1998; Heine et al. 2004). Exercise produces beneficial effects on cognitive function in healthy older adults due to increase in exercise-mediated mitochondrial biogenesis and quality control, including up-regulation of mitophagy (Bori et al. 2012; Lanza and Sreekumaran 2010; Colcombe and Kramer 2003; Erickson and Kramer 2009). Thus, the stability and regulation of mitochondrial dynamics is critical for maintaining neuronal health, connectivity and function. Exercise not only facilitates neuroprotection and neuroplasticity (Stranahan et al. 2009), but also improves physical and cognitive function (Hillman et al. 2008). Exercise also promotes improvements in cognitive function through increase in gray matter volume (Colcombe et al. 2003; Colcombe et al. 2006; Leckie et al. 2012) and neurogenesis in the dentate gyrus. Neurogenesis is known to be coupled with angiogenesis (Palmer et al. 2000; Louissaint et al. 2002), which in turn related to cerebral blood volume (Van Praag et al. 1999). It is hypothesized that measurement of cerebral blood volume may provide an in vivo correlation between neurogenesis and increased cerebral blood flow due to exercise. At the molecular level many molecules mediate exercise-induced neurogenesis, angiogenesis, and cerebral blood flow. These molecules include VEGF (Louissaint et al. 2002) BDNF (Neeper et al. 1996), apolipoprotein E (APOE) (Schuit et al. 2001), catechol-O-methyltransferase (COMT) (Stroth et al. 2010), endorphins, and nitric oxide (Carmargo et al. 2013). This hypothesis is based on the observations that exercise increases both neurogenesis and angiogenesis in young adult rodents (van Praag et al. 2005). Aerobic exercise is not only known to improve brain blood flow, but also increases hippocampal volumes and neuroplasticity (Erickson et al. 2009; Swain et al. 2003; Pereira et al. 2007) supporting the view that improvement in hippocampal blood flow may be closely associated with memory formation, cognitive function, and functional connectivity (Colcombe et al. 2004; Voss et al. 2010). Exercise has been reported to increase the expression of synaptic plasticity genes, gene products such as synapsin I and synaptophysin, and various neuroplasticity-related transcription factors such as cyclic adenosine monophosphate response element binding and intracellular kinases (Stranahan et al. 2010; Vaynman et al. 2004; Vaynman et al. 2006). Exercise also modulates gene involved in insulin-like signaling, energy metabolism, neurogenesis, and synaptic plasticity along with learning and memory (Reagan 2007; Praag et al. 2005; Benedict et al. 2004). Although, the molecular mechanism by which exercise modulates insulin signaling in brain cells is not fully understood, but based on lifelong running studies in rats, it is proposed that MAP kinase and Wnt signaling may contribute to hippocampal plasticity, neurogenesis and learning and memory (Reichardt 2006; Sweatt 2004; Stranahan et al. 2010).
Related posts:
 Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Neurochemical Aspects of Oxidative and Nitrosative Stress
Contribution of Dietary Carbohydrates in Induction of Oxidative Stress
Contribution of Receptors, Transcription Factors, and Genes in the Induction of Neuroinflammation
Contribution of Dietary Fat in Neuroinflammation
Biochemical Aspects of Neuroinflammation
Effect of Exercise on Neurodegeneration in Neurological Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


